Input File: Fluids¶
Relativistic Cold Fluid¶
The following object generates a module that computes the motion of a cold, relativistic, electron fluid. It is assumed there is an immobile background of ions. Temperature only comes into the computation of the Coulomb collision frequency. The electron temperature is controlled by the last generate block (see Matter Loading). There can only be one cold fluid module in a simulation. Electron, ion, and neutral density are tracked in this one module. Any profiles installed for the fluid refer to a gas with constant fractional ionization.
- new fluid <name> { <directives> }
- Parameters:
name (str) – assigns a name to the fluid
directives (block) –
The following directives are supported:
Installable tools: PhotoIonization
- charge = q
- Parameters:
q (float) – charge of the fluid constituent, usually electrons
- mass = m
- Parameters:
m (float) – mass of the fluid constituent, usually electrons
- neutral cross section = sigma
- Parameters:
sigma (float) – electron-neutral collision cross section
- coulomb collisions = cc
- Parameters:
cc (bool) – if true, add collision frequency based on Coulomb cross section to neutral collision frequency
Note
the ion species and electron species variables (see PhotoIonization) have no meaning here
SPARC Hydro Modules¶
- new hydrodynamics [<name>] { <directives> }
This is the top level SPARC module. Internally it contains and manages all other SPARC modules.
- Parameters:
name (str) – name given to the hydro manager
directives (block) –
The following directives are supported:
Installable tools: Particle Movers
- epsilon factor = eps
- Parameters:
eps (float) – error tolerance for adaptive time step
- background density = n0
- Parameters:
n0 (float) – automatically create a uniform background density
n0for every chemical species. Charged species are automatically weighted such that neutrality is maintained. Defaults to zero, in which case the user is responsible for explicitly loading every chemical species.
- background temperature = T0
- Parameters:
T0 (float) – temperature to use for the background fluid. If the background density is nonzero this must be set to a positive value.
- radiation model = rad
- Parameters:
rad (enum) – takes values
thick,thin,none
- laser model = las
- Parameters:
las (enum) – takes values
vacuum,isotropic
- plasma model = plas
- Parameters:
plas (enum) – takes values
neutral,quasineutral
- electrostatic heating = esh
- Parameters:
esh (bool) – If
on, electrons are heated by self-consistent electrostatic fields. Default isoff.
- dipole center = Dx Dy Dz
Reference point for dipole moment diagnostic
- new chemical [<name>] [for <name>] { <directives> }
The
chemicalmodule uses automatic attachment, i.e., if achemicalis created at the root level without theforclause a new equilibrium group module is created for it automatically. As a result,chemicalmodules should never be attached usinggetstatements.- Parameters:
name (str) – name given to the chemical species
directives (block) –
The following directives are supported:
Installable tools: PhotoIonization, Equation of State Tools
- mass = m0
- Parameters:
m0 (float) – mass of the constituent particles, default = 1.0
- charge = q0
- Parameters:
q0 (float) – charge of the constituent particles, default = -1.0
- cv = cv0
- Parameters:
cv0 (float) – normalized specific heat at constant volume,
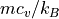 , a typical value is 1.5 for species with no internal degrees of freedom.
, a typical value is 1.5 for species with no internal degrees of freedom.
- vibrational energy = epsv
- Parameters:
epsv (float) – energy between vibrational levels, default = 0 = no vibrations
- implicit = tst
- Parameters:
tst (bool) – whether to use implicit electron advance for this species
- thermometric conductivity = k
- Parameters:
k (float) – Thermometric conductivity. Thermometric conductivity is
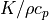 , where K = heat conductivity. For air, k = 2e-5 m^2/s = 0.2 cm^2/s, and K = 2.5e-4 W/(cm*K). SPARC solves the heat equation
, where K = heat conductivity. For air, k = 2e-5 m^2/s = 0.2 cm^2/s, and K = 2.5e-4 W/(cm*K). SPARC solves the heat equation 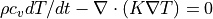 . For electrons the Braginskii conductivity is used.
. For electrons the Braginskii conductivity is used.
- kinematic viscosity = x
- Parameters:
x (float) – Kinematic viscosity. Kinematic viscosity is
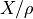 , where X = dynamic viscosity. For air, kinematic viscosity is about 0.15 cm^2/s. SPARC solves the momentum diffusion equation
, where X = dynamic viscosity. For air, kinematic viscosity is about 0.15 cm^2/s. SPARC solves the momentum diffusion equation 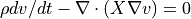 .
.
- effective mass = meff
- Parameters:
meff (float) – effective mass for density >> 1.0 for electrons moving through this chemical
- permittivity = (epsr,epsi)
- Parameters:
epsr (float) – real part of permittivity relative to free space permittivity
epsi (float) – imaginary part of permittivity relative to free space permittivity
- new group [<name>] [for <name>] { directives }
Create an equilibrium group module. This is a container for chemical species that are assumed to be in equilibrium with one another, and therefore have a common temperature and velocity. All chemicals are part of a group.
The
groupmodule uses automatic attachment, i.e., if created at the root level without theforclause it is automatically attached tohydrodynamics. Thehydrodynamicsmodule is automatically created, if necessary.- Parameters:
name (str) – name given to the group
directives (block) –
The following directives are supported:
- mobile = tst
- Parameters:
tst (bool) – whether chemicals in this group are mobile or immobile
SPARC Collision Directives¶
SPARC collisions broadly include elastic and inelastic collisions, as well as chemical reactions. All such processes have to explicitly resolved. These directives are special in that they use a compact, ordered declaration (without the usual parameter block), and use dimensional numbers in CGS-eV units. This is due to the potentially large number of such constructs that may appear in an input file. Dimensional numbers should not be used.
SPARC collision directives should appear at the root level in the input file. They find their parent modules automatically. This makes it more straightforward to #include reaction data from separate files.
- new collision = sp1 <-> sp2 , cross section = sigma
- Parameters:
sp1 (str) – name of chemical species 1 in two-body collision
sp2 (str) – name of chemical species 2 in two-body collision
sigma (float) – cross section in square centimeters
- new collision = sp1 <-> sp1 , coulomb
Uses the Coulomb collision cross section, derived from local conditions.
- Parameters:
sp1 (str) – name of chemical species 1 in two-body collision
sp2 (str) – name of chemical species 2 in two-body collision
- new collision = sp1 <-> sp1 , metallic , ks = ks0 , fermi_energy_ev = ef , ref_density = nref
Uses the harmonic mean of electron-phonon and coulomb collision rates
- Parameters:
ks0 (float) – dimensionless, see K. Eidmann et al., Phys. Rev. E 62, 1202 (2000)
ef (float) – the Fermi energy in eV
nref (float) – the density at which the formula directly applies in particles per cubic centimeter
- new reaction = { eq1 : eq2 : eq3 : ... } rate = c0 c1 c2 cat(range)
Sets up a chemical reaction between arbitrary species using a modified Arrhenius rate
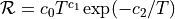
over a range of temperatures. Piecewise rate constructions can be created by using multiple reactions which have the same equation but different rates and different temperature ranges.
- Parameters:
eq1 (str) – chemical equation, or subreaction, in the form
r1 + r2 + ... -> p1 + p2 + ... + eps, wherer1etc. are replaced by names of reactants,p1etc. are replaced by names of products, andepsis a heat of reaction in eV. Breaking the reaction into subreactions can be used to control the flow of energy from reactants to products. The heat of reaction, if negative, is taken from the equilibrium group of the last reactant listed. If positive, it is added to the equilibrium group of the last product listed.c0 (float) – rate coefficient in cm^(3(N-1))/s, where N is the number of reactants
c1 (float) – dimensionless exponent in rate law
c2 (float) – temperature reference appearing in rate law in eV
cat (str) – name of the chemical to be considered the catalyst, i.e., the one whose temperature affects the rate
range (numpy_range) – range of temperatures specified as in numpy, i.e., T1:T2, where T1 and T2 are floating point literals, given in eV. Also as in numpy, :T2 means 0-T2, while T1: means T1-infinity.
- new reaction = { eq1 : eq2 : eq3 : ... } janev_rate = c0 c1 c2 c3 c4 c5 c6 c7 c8 cat(range)
Alternative way of specifying the reaction rate:
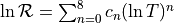
- new excitation = sp1 -> sp2 level = n rate = c0 c1 c2
Vibrational excitation of one species by another. If level = n the transition is between ground and level n. If level = 0 the transition is between adjacent levels, where it is assumed the rate for transitions from n to n+1 is the same for all n.
- new excitation = sp1 -> sp2 level = n janev_rate = c0 c1 c2 c3 c4 c5 c6 c7 c8
Alternative way of specifying the excitation rate.